The main objective of our research is to understand the cellular and molecular mechanisms underlying immunological disease initiation, progression and chronicity, and to provide innovative solutions towards their diagnosis, prognosis and therapy. Towards that end, we use a unique collection of transgenic mouse models, conditional gene targeting, and state-of-the-art cellular and molecular technologies. We combine omics approaches and large-scale data analyses with in depth dissection of candidate gene function in vivo and in vitro, from human vs animal models, to discover causalities in the epigenetic, transcriptional and proteomic changes that occur in specific cell types of interest during disease course. Through the identification and validation of unexplored pathways, targets and biomarkers, we aim to introduce novel concepts towards disease prognosis, prevention and therapy.
Our research team is organized into five groups which are focused on specific organs and diseases or on the development of preclinical therapeutic approaches:
Chronic Inflammatory Arthritis
Inflammatory Bowel Disease (IBD) and Cancer
Neuroinflammation and neuroimmune interactions
Dynamics of Mesenchymal Cells in Lymphoid Microenvironments
Chronic Inflammatory Arthritis
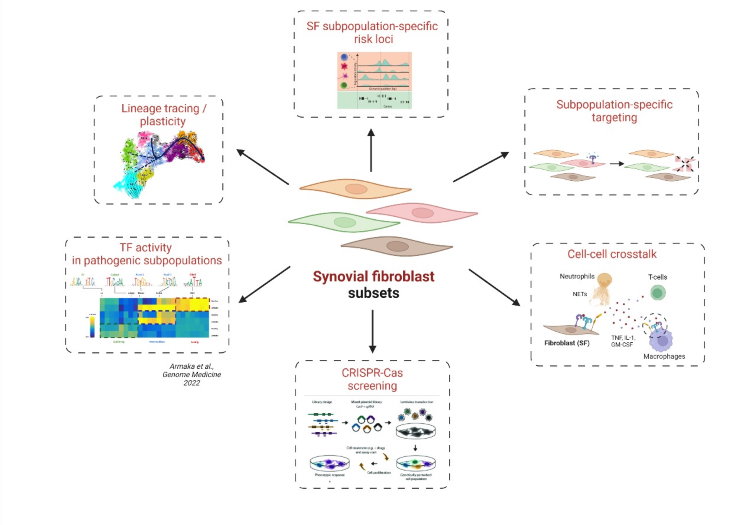
Rheumatoid arthritis (RA) is the most common chronic inflammatory joint disease debilitating 1-2% of the human population. While the exact mechanisms leading to RA pathogenesis remain elusive, experimental evidence from our lab indicates that chronic innate immune activation of the synovial fibroblast (SF) in TNF-overexpressing mice5, is an early event leading to synovial hyperplasia and inflammation. SFs are resident mesenchymal cells of the synovial membrane showing key disease promoting pro-inflammatory activities. While until recently such non-immune cells were regarded as merely targets of the inflammatory microenvironment, and thus secondary effectors, recent evidence suggests that SFs may be the inciting players in RA aetiopathogenesis4,2.
Our current working hypothesis is that chronic activation signals targeting the SFs lead to persistent alterations in epigenetic and gene expression programs, transforming them into an autonomous and aggressive effector cell type inciting the whole spectrum of chronic pathogenic progression in arthritis3. Based on the hypothesis that arthritogenic signals are sustained by multiple layers of epigenetic regulation, we are exploring the impact of regulatory RNAs, such as miRNAs and IncRNAs, working from both ends: a) by using global omics approaches and b) through the development of mouse genetic tools for their in vivo study. Analysis of newly generated scRNA-Seq and scATAC-Seq data in SFs from TNF-overexpressing mice has shed light on the emergence and expansion of pathogenic stroma subpopulations in disease. We explored the differentiation trajectories, that SF subpopulations undergo during arthritis initiation and progression and identified groups of known (NF-kB) and novel transcription factors (Runx1, Bach1), that characterize them1. By using novel genome engineering (in vivo CRISPR/Cas) and bioinformatics tools, our future work focuses on subpopulation-specific targeting of SFs in the synovium, uncovering cross-talk between SFs and neighboring cells and determining key factors, that promote transition from health to disease state.
Selected publications
1. Roumelioti F, Tzaferis C, Konstantopoulos D, Papadopoulou D, Prados A, Sakkou M, Liakos A, Chouvardas P, Meletakos T, Pandis Y, Karagianni N, Denis MC, Fousteri M, Armaka M, Kollias G. (2024) Mir221/222 drive synovial hyperplasia and arthritis by targeting cell cycle inhibitors and chromatin remodeling components. Elife. 13:e84698.
2. Armaka M, Konstantopoulos D, Tzaferis C, Lavigne MD, Sakkou M, Liakos A, Sfikakis PP, Dimopoulos MA, Fousteri M, Kollias G. (2022) Single-cell multimodal analysis identifies common regulatory programs in Synovial Fibroblasts of Rheumatoid Arthritis patients and modelled TNF-driven arthritis. Genome Med., 14(1):78.
3. Armaka M, Ospelt C, Pasparakis M, Kollias G. (2018) The p55TNFR-IKK2-Ripk3 axis orchestrates arthritis by regulating death and inflammatory pathways in synovial fibroblasts. Nat Commun. 12;9(1):618.
4. Pandis I, Ospelt C, Karagianni N, Denis M, Reczko M, Camps C, Hatzigeorgiou A, Ragoussis J, Gay S and Kollias G. (2012) Identification of microRNA-221/222 and -323-3p Association with Rheumatoid Arthritis via Predictions using the Human TNF Transgenic Mouse Model. Ann Rheum Dis. 71(10):1716-23.
5. Armaka M. Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. (2008) Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 18;205(2):331.
6. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 10(3):387-98.
Inflammatory Bowel Disease (IBD) and Cancer
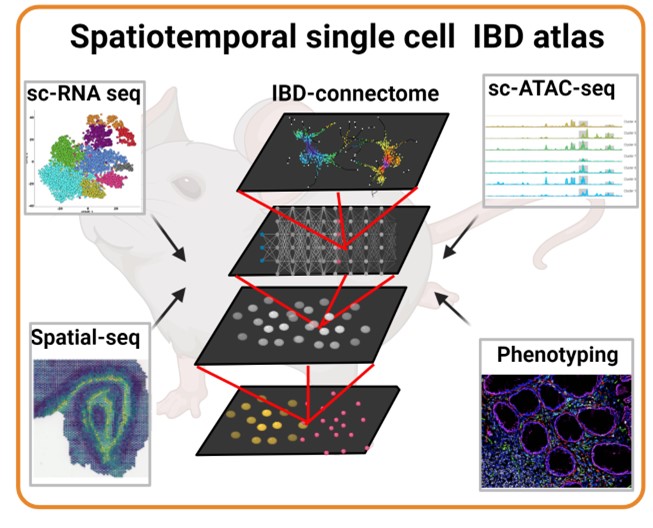
Our previous research work has fundamentally contributed to the establishment of intestinal mesenchymal cells as crucial regulators of intestinal immunity, inflammation and carcinogenesis.1-7 Importantly, we have provided direct evidence for the causal involvement of IMCs in the development of intestinal inflammation.7 More recently, we proposed that in the Peyers Patches (PPs), the secondary lymphoid organs of the small intestine, different mesenchymal populations arise from distinct precursors associated with different anatomic locations, dynamics and molecular requirements.6 This finding introduces a unifying hypothesis, where fibroblast localization and communication cellular signals determine tissue function. Thus, we aim to delineate the dynamic and contextual processes during chronic intestinal inflammation and discover the molecular thresholds and switches that instruct fibroblasts to ‘become’ either resolving, chronic inflammatory or fibrogenic.
To map dynamic chromatin and gene expression programs that define cellular heterogeneity. we are currently using a variety of intestinal inflammation animal models with distinct pathophysiological features, and a collection of mesenchymal specific-cre tools. We are also employing multiplex imaging and spatial sequencing technologies in different disease states to provide novel insights into the spatiotemporal organization of the different fibroblast subsets in the diseased intestine. This integrated approach can enable the identification of fibroblast subset-specific topological niches that interact with the inflammatory microenvironment and change dynamically throughout the disease. In parallel, we are analysing the origin, plasticity, and lineage trajectories of intestinal fibroblasts to perform discovery screens and functional validations on novel fibroblast-subset-specific pathways that may regulate the transition from chronic intestinal inflammation to fibrosis
Finally, in collaboration with the Medical School of Athens, we employ clinical material to validate the involvement of the most prominent new mesenchymal pathways in human disease. To better assess the cellular complexity of human disease we are also involved in the implementation of more complex ex vivo intestinal multicellular systems. Our labs work on the intestinal pathophysiology is currently supported by an Advanced ERC grant 2022-2027, titled: ‘Becoming Causal: Contextual specification of fibroblast-driven causalities in chronic intestinal inflammation and fibrosis’.
Selected publications
1. Iliopoulou L, Tzaferis C, Prados A, Roumelioti F, Koliaraki V, Kollias G. (2025) Different fibroblast subtypes propel spatially defined ileal inflammation through TNFR1 signalling in murine ileitis. Nat Commun. 16(1):3023.
2. Iliopoulou L, Lianopoulou E, Kollias G. (2024) IL-23 exerts dominant pathogenic functions in Crohn's disease-ileitis. Mucosal Immunol. S1933-0219(24)00049-7.
3. Koliaraki, V., Prados, A., Armaka, M. & Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. (2020) doi:10.1038/s41590-020-0741-2.
4. Koliaraki, V., Roulis, M. & Kollias, G. Tpl2 regulates intestinal myofibroblast HGF release to suppress colitis-associated tumorigenesis. J. Clin. Invest. 122, 4231–4242 (2012).
5. Roulis, M. et al. Intestinal myofibroblast-specific Tpl2-Cox-2-PGE2 pathway links innate sensing to epithelial homeostasis. Proc. Natl. Acad. Sci. U. S. A. 111, E4658–E4667 (2014).
6. Koliaraki, V., Pasparakis, M. & Kollias, G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 212, 2235–2251 (2015).
7. Henriques, A., Koliaraki, V. & Kollias, G. Mesenchymal MAPKAPK2/HSP27 drives intestinal carcinogenesis. Proc. Natl. Acad. Sci. 115, E5546–E5555 (2018).
8. Prados, A. et al. Fibroblastic reticular cell lineage convergence in Peyer’s patches governs intestinal immunity. Nat. Immunol. 2021 224 22, 510–519 (2021).
9. Armaka, M. et al. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 205, 331–337 (2008).
Neuroinflammation and neuroimmune interactions
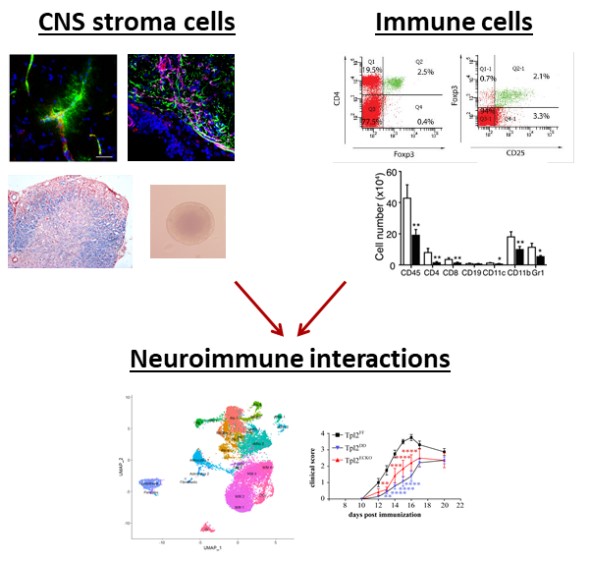
Previous work from our lab has been instrumental in understanding the role of Tumour Necrosis Factor (TNF) and its receptors in neuroinflammation showing the dual TNF function according to receptor binding (1). Importantly, our studies managed for the first time to explain the detrimental failure of anti-TNF therapy in Multiple Sclerosis (MS) patients (1) and provided mechanistic insights on TNF signaling through TNFR1 and TNFR2 (2,3). Further studies focusing on the cellular source and targets of TNF, TNFR1 and TNFR2 have shown that TNFR2 expression on non-hematopoietic cells is required for regulatory T cell function and immune suppression during neuroinflammation (4). Thus, we hypothesised that CNS resident cells play a major role during neuroinflammation and particularly during the resolution of inflammation and CNS regeneration. This was further supported by identifying a novel role of Tpl2, a MAP3-kinase critically involved in the immune response, not in immune cells, but in CNS resident endothelial cells. Specifically, we showed that Tpl2 expression on endothelial cells promotes neuroinflammation through the regulation of vascular permeability and immune cells (or cancer cells) influx (5). Our current studies aim to delineate the interactions of CNS resident cells and immune cells, as well as to identify the spatiotemporal dynamics that regulate remyelination and CNS regeneration.
To further explore the role of CNS resident cells in the regulation of inflammation onset and resolution, we have generated conditional knock out animals for TNFR2 and Tpl2 allowing the cell-specific ablation of the proteins. We use different models of neuroinflammation and demyelination (such as Experimental Autoimmune Encephalomyelitis, LPS, Cuprizone) to explore neuroimmune interactions at different stages of pathology to understand the spatiotemporal organization of the CNS through single-nuclei RNA-seq and spatial proteomics experiments. To validate our findings and gain mechanistic insight, we use in vitro models (co-cultures), while we are currently developing 3D culture models. We are also planning to generate additional transgenic mouse lines through the CRISPR-Cas9 system to fully characterize the dynamics of remyelination and CNS regeneration with ultimate aim the induction of these processes in vivo.
Selected publications
1. Kassiotis, G. and Kollias, G. (2001) Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. The Journal of experimental medicine, 193, 427-434.
2. Akassoglou, K., Douni, E., Bauer, J., Lassmann, H., Kollias, G. and Probert, L. (2003) Exclusive tumor necrosis factor (TNF) signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 100, 709-714.
3. Alexopoulou, L., Kranidioti, K., Xanthoulea, S., Denis, M., Kotanidou, A., Douni, E., Blackshear, P.J., Kontoyiannis, D.L. and Kollias, G. (2006) Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. European journal of immunology, 36, 2768-2780.
4. Tsakiri, N., Papadopoulos, D., Denis, M.C., Mitsikostas, D.D. and Kollias, G. (2012) TNFR2 on non-haematopoietic cells is required for Foxp3+ Treg-cell function and disease suppression in EAE. European journal of immunology, 42, 403-412.
5. Nanou, A., Bourbouli, M., Vetrano, S., Schaeper, U., Ley, S. and Kollias, G. (2021) Endothelial Tpl2 regulates vascular barrier function via JNK-mediated degradation of claudin-5 promoting neuroinflammation or tumor metastasis. Cell reports, 35, 109168.
Dynamics of Mesenchymal Cells in Lymphoid Microenvironments
The adaptive immune response is initiated in secondary lymphoid organs (SLOs), including lymph nodes, spleen and Peyer’s patches in the intestine. These organs act as elaborate filters, located in strategic sites to maximize the chance of an encounter between lymphocytes and antigens. Despite their different macroscopic structure, they all share a complex microanatomy and the common feature of lymphocyte segregation in two different compartments, the T- and B-cell area. Behind this compartmentalization lies a heterogeneous population of mesenchymal cells (MCs) that orchestrates the formation and maintenance of SLO structure and regulates the migration and function of immune cells.
Traditionally, at least three major mesenchymal populations have been described in all SLOs: marginal reticular cells and follicular dendritic cells (FDCs) in the B cell area and the fibroblastic reticular cells in the T cell area. Yet, recent studies using single-cell RNA sequencing revealed a more heterogeneous scenario. We have previously demonstrated that the development of FDC networks requires the expression of TNFR1 on CD21-Cre+ precursors. 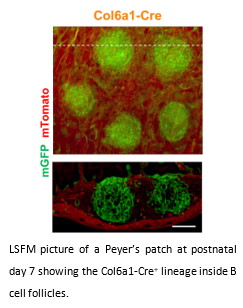 However, the ontogenetic relations between FDCs and the other MC populations, as well as the molecular circuits governing their differentiation pathways have not been clearly established yet.
However, the ontogenetic relations between FDCs and the other MC populations, as well as the molecular circuits governing their differentiation pathways have not been clearly established yet.
Our current working hypothesis is that SLO MC subpopulations arise from distinct precursors associated with different anatomic locations, dynamics and molecular requirements. To verify this hypothesis, we use Cre driver mouse lines with different MC specificities in combination with multicolor fate mapping systems and light sheet fluorescent microscopy (LSFM). This approach allows us to characterize new mesenchymal lineages throughout the development of Peyer's patches. We are also exploring the effect of lineage-specific gene ablation on Peyer’s patch differentiation and its impact on the immune system and the intestinal microbiota composition.
Selected publications
1. Victoratos P, Kollias G. (2009) Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity. 30(1):130-42.
2. Victoratos P, Lagnel J, Tzima S, Alimzhanov MB, Rajewsky K, Pasparakis M, Kollias G. (2006) FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity. 24(1):65-77.
3. Pasparakis M, Alexopoulou L, Grell M, Pfizenmaier K, Bluethmann H, Kollias G. (1997), Peyer's patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor, Proc Natl Acad Sci U S A. 94(12):6319-6323.
Drug Development Research
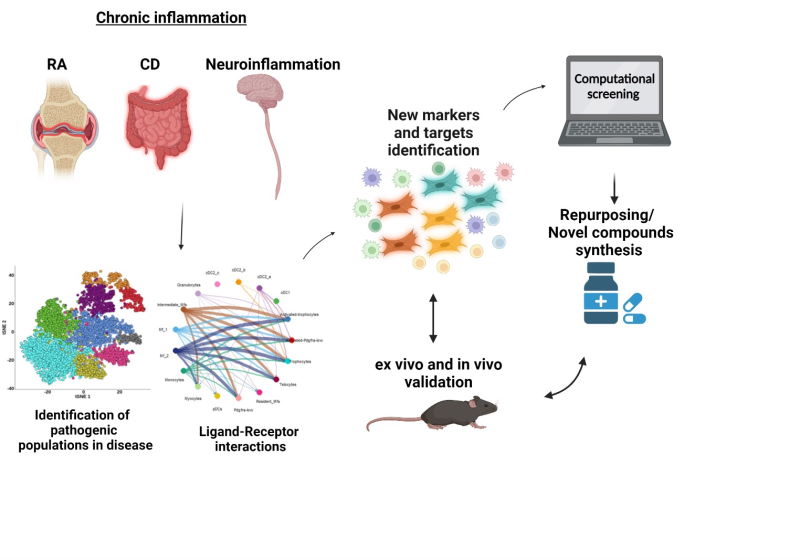
The drug development research group is an integrated part of our team, bridging the gap between basic and clinical research. The aim of this group is to further the potential of our research projects through the identification of drug candidates and novel therapeutic targets involved in the pathogenesis of chronic inflammatory diseases. We work on early drug discovery to establish novel translational pipelines that include target identification and validation, assay development, lead optimization and preclinical testing. Towards that end, we use a unique collection of transgenic mouse models and work together with the medicinal chemistry group at BSRC Fleming and our spin-off company Biomedcode. Through the identification and validation of unexplored pathways and targets we aim to introduce novel concepts towards disease therapy.
The past years, our group has focused on the development of novel drugs for the treatment of rheumatoid arthritis (RA) by using a phenotypic assay approach. As we have previously shown, chronic overexpression of human TNF (hTNF) from joint synovial fibroblasts (SFs) is sufficient to provoke RA in transgenic mice (hTNFtg). Based on the gene expression profile of hTNFtg fibroblasts, we have repurposed an FDA approved neuroleptic drug, which was validated to reduce SFs’ inflammatory potential, while decreasing the clinical score of hTNFtg polyarthritis. Based on the core structure of this compound, we synthesize novel small molecule derivatives – as lead compounds - which are evaluated for their ability to inhibit pathogenic activation of synovial fibroblasts and for their drug-like properties. The aforementioned studies are supported by Drug.Art (Τ2ΕΔΚ-01076) funding of General Secretariat for Research and Innovation, while novel compounds are patented under #PCT/EP2022/056349.
Integrating single-cell transcriptomics studies of mouse models of RA, Crohn’s disease and neuroinflammation with relevant available human data, we now focus on investigating a. unique markers that characterize pathogenic fibroblasts’ clusters and b. ligand-receptor cellular interactors, aiming to identify novel potential therapeutic targets involved in the pathogenesis of chronic inflammation. Potential inhibitors of these targets, will be further studied for their mechanism of action and their therapeutic potential in animal models of chronic inflammation.
The originality of our work focuses on: a) the targeting of a cell type (mesenchymal cells) that to date has not been exploited therapeutically, even though it has been shown to play a key role in the pathogenesis of chronic inflammatory diseases, b) the discovery of novel small molecules as potential therapeutics in high demand from the market as successors of current biological treatments.
Selected publications
1. Papadopoulou D, Mavrikaki V, Charalampous F, Tzaferis C, Samiotaki M, Papavasileiou KD, Afantitis A, Karagianni N, Denis MC, Sanchez J, Lane JR, Faidon Brotzakis Z, Skretas G, Georgiadis D, Matralis AN, Kollias G. (2024) Discovery of the First-in-Class Inhibitors of Hypoxia Up-Regulated Protein 1 (HYOU1) Suppressing Pathogenic Fibroblast Activation. Angew Chem Int Ed Engl. 63(14):e202319157.
2. Papadopoulou D, Roumelioti F, Tzaferis C, Chouvardas P, Pedersen AK, Charalampous F, Christodoulou-Vafeiadou E, Ntari L, Karagianni N, Denis MC, Olsen JV, Matralis AN, Kollias G. (2023) Repurposing antipsychotic drug Amisulpride for targeting synovial fibroblast activation in arthritis. JCI Insight. 8(9):e165024.
3. Papadopoulou, D. et al. In silico identification and evaluation of natural products as potential tumor necrosis factor function inhibitors using advanced enalos asclepios KNIME nodes. Int. J. Mol. Sci. (2021).
4. Ntari, L. et al. Combination of subtherapeutic anti-TNF dose with dasatinib restores clinical and molecular arthritogenic profiles better than standard anti-TNF treatment. J Transl Med. (2021).
5. Karagianni, N. et al. An integrative transcriptome analysis framework for drug efficacy and similarity reveals drug-specific signatures of anti-TNF treatment in a mouse model of inflammatory polyarthritis. PLoS Comput. Biol. (2019).
6. Melagraki, G. et al. In silico discovery of plant-origin natural product inhibitors of tumor necrosis factor (TNF) and receptor activator of NF-κB ligand (RANKL). Front. Pharmacol. (2018).
7. Melagraki, G. et al. Cheminformatics-aided discovery of small-molecule Protein-Protein Interaction (PPI) dual inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-κB Ligand (RANKL). PLoS Comput. Biol. (2017).