Elucidation of RANKL-mediated pathogenic mechanisms and identification of novel RANKL inhibitors
 Receptor Activator of Nuclear Factor-κB Ligand (RANKL) is the primary mediator of bone loss through osteoclast-induced bone resorption. RANKL binds as a trimer to its receptor RANK that is expressed on the surface of osteoclast precursors initiating a signaling cascade that results in osteoclast differentiation which leads to bone resorption. Disruption of RANKL function leads to recessive osteopetrosis due to failure of osteoclast formation whereas overproduction of RANKL is implicated in a variety of degenerative bone diseases such as osteoporosis. A fully human monoclonal antibody against RANKL has been recently approved for the treatment of patients with osteoporosis and in prostate cancer patients undergoing hormonal ablation therapy. Therefore, RANKL is considered the major therapeutic target for the suppression of bone resorption in bone metabolic diseases such as osteoporosis, rheumatoid arthritis and cancer metastasis. Our lab has generated unique models of RANKL-mediated osteoporosis and osteopetrosis and has identified novel RANKL inhibitors.
Receptor Activator of Nuclear Factor-κB Ligand (RANKL) is the primary mediator of bone loss through osteoclast-induced bone resorption. RANKL binds as a trimer to its receptor RANK that is expressed on the surface of osteoclast precursors initiating a signaling cascade that results in osteoclast differentiation which leads to bone resorption. Disruption of RANKL function leads to recessive osteopetrosis due to failure of osteoclast formation whereas overproduction of RANKL is implicated in a variety of degenerative bone diseases such as osteoporosis. A fully human monoclonal antibody against RANKL has been recently approved for the treatment of patients with osteoporosis and in prostate cancer patients undergoing hormonal ablation therapy. Therefore, RANKL is considered the major therapeutic target for the suppression of bone resorption in bone metabolic diseases such as osteoporosis, rheumatoid arthritis and cancer metastasis. Our lab has generated unique models of RANKL-mediated osteoporosis and osteopetrosis and has identified novel RANKL inhibitors.
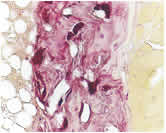 RANKL-mediated osteoporosis: We have recently generated transgenic mice overexpressing human RANKL (TghuRANKL) in order to model human RANKL-mediated pathologies. Two transgenic lines were generated: (i) the low copy number Tg5516 line that develops a mild osteoporotic phenotype characterized by trabecular bone loss and reduced biomechanical properties, and (ii) the high copy Tg5519 line that exhibit a severe osteoporotic phenotype with trabecular bone loss, severe cortical bone porosity, increased serum markers of bone turnover and decreased bone strength. The observed phenotypes develop both in males and females, and the levels of human RANKL expression are correlated with bone resorption and disease severity. Our ongoing studies are focused on osteoimmunology by elucidating the role of RANKL in osteoporosis and inflammatory bone diseases. Transgenic mice expressing human RANKL represent a unique tool for understanding the pathogenic mechanisms that cause bone resorption and for the evaluation of novel therapeutic approaches targeting RANKL-mediated pathologies such as osteoporosis.
RANKL-mediated osteoporosis: We have recently generated transgenic mice overexpressing human RANKL (TghuRANKL) in order to model human RANKL-mediated pathologies. Two transgenic lines were generated: (i) the low copy number Tg5516 line that develops a mild osteoporotic phenotype characterized by trabecular bone loss and reduced biomechanical properties, and (ii) the high copy Tg5519 line that exhibit a severe osteoporotic phenotype with trabecular bone loss, severe cortical bone porosity, increased serum markers of bone turnover and decreased bone strength. The observed phenotypes develop both in males and females, and the levels of human RANKL expression are correlated with bone resorption and disease severity. Our ongoing studies are focused on osteoimmunology by elucidating the role of RANKL in osteoporosis and inflammatory bone diseases. Transgenic mice expressing human RANKL represent a unique tool for understanding the pathogenic mechanisms that cause bone resorption and for the evaluation of novel therapeutic approaches targeting RANKL-mediated pathologies such as osteoporosis.
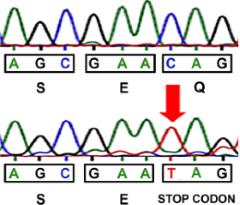
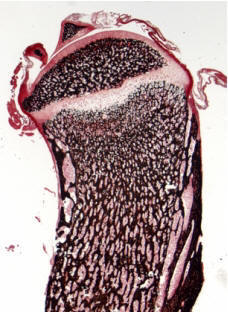 Identification of novel RANKL inhibitors: We have recently identified a recessive loss-of-function Rankl allele in Rankltles/tles mice causing severe osteopetrosis, failure of tooth eruption and defective lymph node organogenesis similarly to Rankl-deficient mice. The causal missense mutation contributes to a single aminoacid substitution at the extracellular domain of RANKL that inhibits trimer assembly exerting also a dominant negative effect. Based on the RANKL crystal structure and the position of the conserved functional aminoacid we have identified novel small molecules that inhibit RANKL function using ex vivo osteoclastogenesis assays. Such inhibitors are currently being characterized biochemically and are being evaluated preclinically in the TgRANKL models of osteoporosis.
Identification of novel RANKL inhibitors: We have recently identified a recessive loss-of-function Rankl allele in Rankltles/tles mice causing severe osteopetrosis, failure of tooth eruption and defective lymph node organogenesis similarly to Rankl-deficient mice. The causal missense mutation contributes to a single aminoacid substitution at the extracellular domain of RANKL that inhibits trimer assembly exerting also a dominant negative effect. Based on the RANKL crystal structure and the position of the conserved functional aminoacid we have identified novel small molecules that inhibit RANKL function using ex vivo osteoclastogenesis assays. Such inhibitors are currently being characterized biochemically and are being evaluated preclinically in the TgRANKL models of osteoporosis.
1. Douni E, Rinotas V, Makrinou E, Zwerina J, Penninger JM, Eliopoulos E, Schett G, Kollias G. (2012). A RANKL G278R mutation causing osteopetrosis identifies a functional amino acid essential for trimer assembly in RANKL and TNF. Human Molecular Genetics; 21(4):784-98.
2. Papaneophytou CP, Mettou AK, Rinotas V, Douni E, Kondopidis GA. (2013). Solvent Selection for Insoluble Ligands, a Challenge for Biological Assay Development: A TNF-α/SPD304 Study. ACS Medicinal Chemistry Letters; 4 (1):137–141.
3. Papaneophytou CP, Rinotas V, Douni E, Kontopidis G. (2013). A statistical approach for optimization of RANKL overexpression in Escherichia coli: Purification and characterization of the protein. Protein Expression and Purification; 90(1):9-19.
4. Rinotas V, Niti A, Dacquin R, Bonnet N, Stolina M, Han CY, Kostenuik P, Jurdic P, Ferrari S, Douni E. (2014). Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. Journal of Bone and Mineral Research; 29(5):1158-69.
5. Alexiou P, Papakyriakou A, Ntougkos E, Papaneophytou CP, Liepouri F, Mettou A, Katsoulis I, Maranti A, Tsiliouka K, Strongilos A, Chaitidou S, Douni E, Kontopidis G, Kollias G, Couladouros E, Eliopoulos E. (2014). Rationally designed less toxic SPD-304 analogs and preliminary evaluation of their TNF inhibitory effects. Arch Pharm; 347(11):798-805.
6. Papaneophytou C, Alexiou P, Papakyriakou A, Ntougkos E, Tsiliouka K, Maranti A, Liepouri F, Strongilos A, Mettou A, Couladouros E, Eliopoulos E, Douni E, Kollias, G, Kontopidis G. (2015). Synthesis and biological evaluation of potential small moleculeinhibitors of tumor necrosis factor. Med. Chem. Commun; 6: 1196-1209.
7. Melagraki G, Ntougkos E, Rinotas V, Papaneophytou C, Leonis G, Mavromoustakos T, Kontopidis G, Douni E, Afantitis A, Kollias G. (2017). Cheminformatics-aided discovery of small molecule Protein-Protein Interaction (PPI) dual inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-κB Ligand (RANKL). PLoS Comput Biol; 13(4):e1005372.
8. Rinotas V, Douni E. (2018). Molecular Interaction of BMAT with Bone. Current Molecular Biology Reports; 4(2):34–40.(Review).
9. Melagraki G, Leonis G, Ntougkos E, Rinotas V, Papaneophytou C, Mavromoustakos T, Kontopidis G, Douni E, Kolias G, Afantitis A. (2018). Current Status and Future Prospects of Small–molecule Protein–protein Interaction (PPI) Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-κB Ligand (RANKL). Curr Top Med Chem; 18(8):661-673. (Review).
10. Melagraki G, Ntougkos E, Papadopoulou D, Rinotas V, Leonis G, Douni E, Afantitis A, Kollias G. (2018). In Silico Discovery of Plant-Origin Natural Product Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-κB Ligand (RANKL). Front Pharmacol; 9:800.
11. Papadaki M, Rinotas V, Violitzi F, Thireou T, Panayotou G, Samiotaki M, Douni E. (2019). New insights for RANKL as a proinflammatory modulator in modeled inflammatory arthritis. Frontiers in Immunology. doi: 10.3389/fimmu.2019.00097.
Identification of a Novel DnaJC Protein Involved in Mitochondrial Structure and Proper Neuromuscular Function
Neuromuscular diseases encompass a wide range of clinical conditions remaining incurable while the genetic and molecular basis of most of these conditions remains unknown. Using mouse ENU mutagenesis we have identified a novel autosomal recessive neuromuscular phenotype characterized by hind limb weakness, progressing with muscle atrophy, reduced body weight, atrophy of lymphoid organs, generalized paralysis and death within the first month of age. The most striking neuropathological finding is the severe vacuolation of the motor neurons in the spinal cord, reminiscent of other mouse models of motor neuron disease. Ultrastructural analysis of motor neuron mitochondria showed that these vacuoles are mitochondria that have disorganized or completely absent cristae. Genetic linkage analysis and sequencing revealed an intronic point mutation in the DnaJC11 gene, a member of the DnaJ homolog, subfamily C (DnaJC) of heat shock proteins (Hsp40s). The intronic mutation generates a novel splicing acceptor site resulting in the insertion of a small intronic region in the mature transcript, causing a replacement of the C terminus of the protein by a new sequence. Genetic confirmation about the causality of this mutation in our phenotype was achieved by complete rescue of the phenotype in the presence of the human DnaJC11 transgene. This is a novel gene with unknown function, whose protein localizes in mitochondria. Our neuromuscular model represents the first evidence that associates mitochondrial structure alterations and the DnaJC family with motor neuron disease pathogenesis. In our current studies we aim to elucidate the molecular basis of the neuromuscular phenotype by identifying the function(s) of this novel DnaJC member.
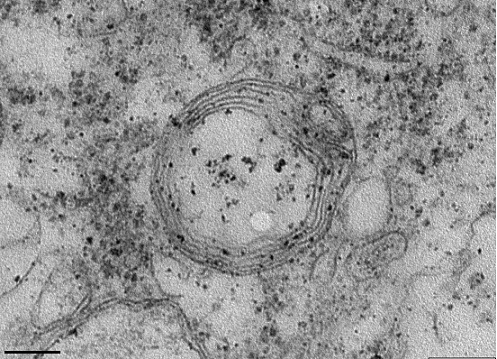
1. Ioakeimidis F, Ott C, Kozjak-Pavlovic V, Violitzi F, Rinotas V, Makrinou E, Eliopoulos E, Fasseas C, Kollias G, Douni E. (2014). A splicing mutation in the novel mitochondrial protein DNAJC11 causes motor neuron pathology associated with cristae disorganization, and lymphoid abnormalities in mice. PLoS One; 9(8):e104237.
Identification of a Novel SLC25 Member Causing Autosomal Recessive Ataxia
SLC25A46, a novel mitochondrial protein, has recently been identified as a pathogenic cause in a wide spectrum of rare human neurological diseases, including optic atrophy, Charcot-Marie-Tooth type 2, Leigh syndrome, progressive myoclonic ataxia and lethal congenital pontocerebellar hypoplasia. Following a genetic approach, our group identified a novel neurological mouse model caused by a nonsense mutation in the Slc25a46 gene. SLC25A46 is an outer membrane protein, member of the Solute Carrier 25 (SLC25) family of nuclear genes encoding mitochondrial carriers, with a role in mitochondrial dynamics and cristae maintenance. Mutant mice manifest the main clinical features identified in patients, including ataxia, optic atrophy and cerebellar hypoplasia, which were completely rescued by expression of the human ortholog. Histopathological analysis revealed previously unseen lesions, most notably disrupted cytoarchitecture in the cerebellum and retina and prominent abnormalities in the neuromuscular junction. A distinct lymphoid phenotype was also evident. Our mutant mice provide a valid model for understanding the mechanistic basis of the complex SLC25A46-mediated pathologies, and further investigation of the associated human pathology.
1. Terzenidou ME, Segklia A, Kano T, Papastefanaki F, Karakostas A, Charalambous M, Ioakeimidis F, Papadaki M, Kloukina I, Chrysanthou-Piterou M, Samiotaki M, Panayotou G, Matsas R, Douni E. (2017). Novel insights into SLC25A46-related pathologies in a genetic mouse model. PLoS Genet; Apr 4;13(4):e1006656. doi: 10.1371.