1. Mechanisms of habituation.
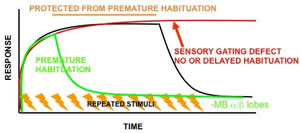 Habituation is a major mechanism to decrease responsiveness to repetitive or prolonged non-reinforced stimuli. Filtering such events of low significance is likely the foundation of selective attention, necessary for learning and memory. Defective habituation has been associated with schizophrenia, learning disabilities such as Attention Deficit Hyperactivity Disorder (ADHD) and migraines among other conditions. We established a system to study habituation to two qualitativel different stimuli and identified and characterized at least one mutant in the process. We are conducting genetic screens aiming to elucidate the molecular basis of habituation for which very little is known and to map neurons within the brain that mediate habituation and protection from premature habituation. We use P and Minos transposons to search for mutations that shorten the period the brain uses to evaluate stimuli (premature habituation-ADHD-like) and others that do not allow habituation to the repeated stimulation. In addition, we are screening drugs likely to reverse the ADHD-like premature habituation of already extant mutants.
Habituation is a major mechanism to decrease responsiveness to repetitive or prolonged non-reinforced stimuli. Filtering such events of low significance is likely the foundation of selective attention, necessary for learning and memory. Defective habituation has been associated with schizophrenia, learning disabilities such as Attention Deficit Hyperactivity Disorder (ADHD) and migraines among other conditions. We established a system to study habituation to two qualitativel different stimuli and identified and characterized at least one mutant in the process. We are conducting genetic screens aiming to elucidate the molecular basis of habituation for which very little is known and to map neurons within the brain that mediate habituation and protection from premature habituation. We use P and Minos transposons to search for mutations that shorten the period the brain uses to evaluate stimuli (premature habituation-ADHD-like) and others that do not allow habituation to the repeated stimulation. In addition, we are screening drugs likely to reverse the ADHD-like premature habituation of already extant mutants.
2. Molecular mechanisms of learning and memory.
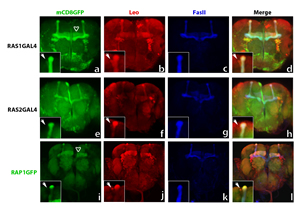 Identification of the molecular components of neuronal signaling cascades and determination of their role in neuroplasticity is essential to understanding learning and memory. To that end, we are employing a multidisciplinary approach based on the knowledge that 5000 specific neurons in the Drosophila brain organized in a characteristic structure called mushroom bodies (MBs) are required for olfactory learning and memory. We have shown that members of the RAS/MAPK signaling cascade expressed in MB neurons when mutated or overexressed precipitate
Identification of the molecular components of neuronal signaling cascades and determination of their role in neuroplasticity is essential to understanding learning and memory. To that end, we are employing a multidisciplinary approach based on the knowledge that 5000 specific neurons in the Drosophila brain organized in a characteristic structure called mushroom bodies (MBs) are required for olfactory learning and memory. We have shown that members of the RAS/MAPK signaling cascade expressed in MB neurons when mutated or overexressed precipitate
deficits, or in certain cases enhancement of learning and long-term memory. This lead us to pursue experimentally the hypothesis that unlike their explored activities in differentiation, proliferation and oncogenicity, members of the cascade have unique roles and regulation in post-developmental neurons mediated via novel interactors or alternative signaling routes. We are focusing on genes with roles in long-term memory formation and stability, as well as the form of consolidated memory known as Anesthesia Resistant. We are also interested in the interaction of signaling cascades with cAMP signaling, which has been established as essential for these processes in multiple experimental systems.
3. Behavioural Neurobiology of Cognitive and Degenerative disorders.
We use transgenic models that express human genes associated with common disorders such as Tauopathies, Alzheimer's and common learning disabilities such as Neurofibromatosis 1 and the Fragile X syndrome to investigate the molecular mechanisms responsible for the associated learning and memory deficits to understand the underlying pathobiology and attempt their pharmacological amelioration eventually.
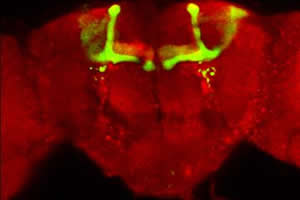
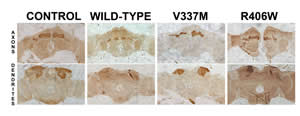 A. Tau-dependent disorders. We established that accumulation of the neuronal microtubule associated protein Tau, akin to that observed in neurons of Frontotemporal Dementia with Parkinsonism and Alzheimer's patients prior to visible degeneration and neurofibrillary tangle (NFT) formation, precipitates large deficits in learning and memory. These defects underlie the dramatic toxicity of Tau on mushroom body neurons shown in the figure. However, using the TARGET system to limit expression to adult mushroom body neurons we can recapitulate the learning deficits without the structural deficits of the MBs, a clear demonstration of adult-specific neuronal dysfunction. Tau-dependent toxicity is still manifested and quantitated as reduced lifespan. We use these two bioassays to investigate the contribution of particular of the 84 phosphorylation sites on Tau to toxicity and dysfunction underlying learning deficits. We are extrapolating our findings to mouse Tauopathy models and human patients.
A. Tau-dependent disorders. We established that accumulation of the neuronal microtubule associated protein Tau, akin to that observed in neurons of Frontotemporal Dementia with Parkinsonism and Alzheimer's patients prior to visible degeneration and neurofibrillary tangle (NFT) formation, precipitates large deficits in learning and memory. These defects underlie the dramatic toxicity of Tau on mushroom body neurons shown in the figure. However, using the TARGET system to limit expression to adult mushroom body neurons we can recapitulate the learning deficits without the structural deficits of the MBs, a clear demonstration of adult-specific neuronal dysfunction. Tau-dependent toxicity is still manifested and quantitated as reduced lifespan. We use these two bioassays to investigate the contribution of particular of the 84 phosphorylation sites on Tau to toxicity and dysfunction underlying learning deficits. We are extrapolating our findings to mouse Tauopathy models and human patients.
Moreover, we are using the fly nervous system and a proteomic to define proteins that interact specifically with the different normal and mutated Tau isoforms to address the hypothesis that these proteins are functionally different and therefore implicated differentially in the different Tauopathies. We are extrapolating our findings to mouse Tauopathy models and human patients.
Finally, we are using genetic and pharmacological approaches to determine the normal function of Tau, its mode of action, regulation and interacting partners in the fly nervous system.
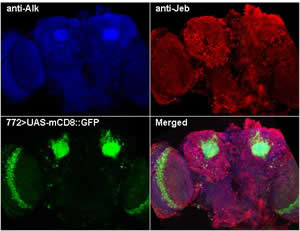 B. Neurofibromatosis 1. Recent evidence from our laboratory indicates that the Receptor Tyrosine Kinase (RTK) Anaplastic Lymphoma Kinase (Alk), which is activated in several cancers, activates the Neurofibromin (Nf1)-regulated signals to Ras in the Drosophila CNS. In fact, in addition to extensive co-localization in the CNS, genetic or pharmacological inhibition of Drosophila Alk (dAlk) rescued the reduced body size,
B. Neurofibromatosis 1. Recent evidence from our laboratory indicates that the Receptor Tyrosine Kinase (RTK) Anaplastic Lymphoma Kinase (Alk), which is activated in several cancers, activates the Neurofibromin (Nf1)-regulated signals to Ras in the Drosophila CNS. In fact, in addition to extensive co-localization in the CNS, genetic or pharmacological inhibition of Drosophila Alk (dAlk) rescued the reduced body size,
adult learning deficits and MAPK overactivation (pMAPK) dNf1mutant phenotypes. Amelioration (but not full reversal), of the dNf1deficient learning phenotypes was achieved with the Alk inhibitor TAE684, strongly suggesting the human RTK as a potential therapeutic target for the cognitive symptoms of NF1 patients. However, one of the challenges of devising treatments for NF1 patients is the extreme variability of their symptoms (phenotypes). One of the overall aims of the experimental approach is to correlate phenotypic variability of NF1 patients with validated molecular aetiologies. Using novel Drosophila mutants and a pharmacogenetic approach we aim to gain insights into this variability of the cognitive symptoms by examining the relationship of ADHD-like premature habituation associative learning and long-term memory in the mutants. Using a combinatorial approach we also aim to fully ameliorate these behavioral defects in two types of mutants, a protein null and a novel point mutation that affects the largely uncharacterized IRA domain of Neurofibromin.
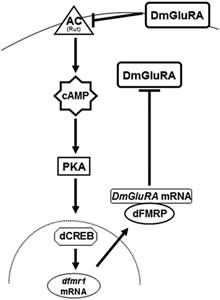 C. Fragile-X syndrome. Fragile X syndrome (FXS) results from loss or functional silencing of the FMR1 gene. The gene encodes the FMR protein (FMRP), involved in mRNA export, localization and translational regulation, especially locally at particular synapses. However, the role of FMRP in these processes has not been defined with certainty and it is therefore difficult to understand the molecular processes and circuits perturbed upon its loss to precipitate the range of behavioral deficits that characterize FXS. It is unclear for example whether the learning deficits and the autism of FXS patients reflect alterations in the same groups of neurons and are functionally related. Individuals with FXS display a range of developmental and behavioral deficits with mild to severe mental retardation, attention deficit disorder and autistic behaviors among others
C. Fragile-X syndrome. Fragile X syndrome (FXS) results from loss or functional silencing of the FMR1 gene. The gene encodes the FMR protein (FMRP), involved in mRNA export, localization and translational regulation, especially locally at particular synapses. However, the role of FMRP in these processes has not been defined with certainty and it is therefore difficult to understand the molecular processes and circuits perturbed upon its loss to precipitate the range of behavioral deficits that characterize FXS. It is unclear for example whether the learning deficits and the autism of FXS patients reflect alterations in the same groups of neurons and are functionally related. Individuals with FXS display a range of developmental and behavioral deficits with mild to severe mental retardation, attention deficit disorder and autistic behaviors among others
For syndromes such as Fragile X Syndrome (FXS), defining the relationship between apparently discordant clinical manifestations, affected neuronal circuits and potentially circuit-specific molecular mechanisms affected is essential. The main objectives of the project is to genetically and pharmacologically investigate the molecular mechanisms perturbed by FMRP loss and the neuronal circuits housing these mechanisms affected by such mutations leading to altered habituation, associative learning and long-term memory. Moreover, we have uncovered novel phenotypes associated with FMRP abrogation of delayed habituation to repetitive non-predictive stimuli reminiscent of the behavioral inflexibility, autism and attention deficits associated with FXS in patients. These studies will be coupled as they have in past with use of pharmacological agents to attempt amelioration of the mutant phenotypes guided by the affected molecules, mechanisms and neuronal circuits revealed in our genetic molecular and behavioral analyses.
4. Molecular and Behavioral Biology of 14-3-3 proteins.
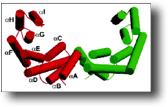 The 14-3-3 proteins comprise a highly conserved family of small acidic molecules present in all eukaryotes. 14-3-3s share a common structure composed of nine anti-parallel α-helices forming a palisade around a central negatively charged groove of largely invariant amino acids. All 14-3-3s form homo- and heterodimers. A phosphoprotein binding surface formed by conserved amino acids in the groove interacts with target proteins that contain the motifs RSxpS/TxP, or RxxxpS/TxP (where x=any amino acid, pS/T=phosphoserine or phosphothreonine). 14-3-3 binding on a target protein may protect it from dephosphorylation or proteolysis, modulate its activity, alter its ability to interact with other partners, or modify its cytoplasmic/nuclear partition. Therefore, it is not unusual that 14-3-3 proteins have been implicated in a diverse number of processes and biochemical pathways.
The 14-3-3 proteins comprise a highly conserved family of small acidic molecules present in all eukaryotes. 14-3-3s share a common structure composed of nine anti-parallel α-helices forming a palisade around a central negatively charged groove of largely invariant amino acids. All 14-3-3s form homo- and heterodimers. A phosphoprotein binding surface formed by conserved amino acids in the groove interacts with target proteins that contain the motifs RSxpS/TxP, or RxxxpS/TxP (where x=any amino acid, pS/T=phosphoserine or phosphothreonine). 14-3-3 binding on a target protein may protect it from dephosphorylation or proteolysis, modulate its activity, alter its ability to interact with other partners, or modify its cytoplasmic/nuclear partition. Therefore, it is not unusual that 14-3-3 proteins have been implicated in a diverse number of processes and biochemical pathways.
Unlike vertebrates which contain seven 14-3-3 genes, Drosophila melanogaster contains two encoding for three 14-3-3ζ proteins proteins (LEO I, LEO II and LEO III) with 88% identity to mammalian isoforms and the single D14-3-3ε protein with 82% identity to its mammalian ortholog. As in vertebrates, the LEO proteins are found enriched in adult brain and in low levels throughout the body, as well as embryos, larvae and pupae. Similarly, D14-3-3ε is present in all developmental stages and in all tissues examined with only slight enrichment in the adult brain. Therefore, Drosophila provides a simple genetically tractable model system to study 14-3-3 isotype specific functions and interactions in vivo. We have determined that the two genes are coordinately regulated in the fly, such that there appeares to be 14-3-3 homeostasis in neurons.
 We are using transgenic animals and tissue and temporal-specific RNAi interference to determine the role of these proteins in habituation, leaning and long-term memory, as well as their putative roles in dopaminergic neurons. In addition we are investigating the functional role of homo and heterodimers in neurons. Finally, we are using a functional genetic approach to determine the conserved functions of vertebrate and other eukaryotic 14-3-3s with respect to supporting viability and normal neuronal function in mutant flies lacking the respective 14-3-3 proteins.
We are using transgenic animals and tissue and temporal-specific RNAi interference to determine the role of these proteins in habituation, leaning and long-term memory, as well as their putative roles in dopaminergic neurons. In addition we are investigating the functional role of homo and heterodimers in neurons. Finally, we are using a functional genetic approach to determine the conserved functions of vertebrate and other eukaryotic 14-3-3s with respect to supporting viability and normal neuronal function in mutant flies lacking the respective 14-3-3 proteins.
5. Detection mechanisms of odorant molecule vibrations. (In collaboration with Dr LUCA TURIN)
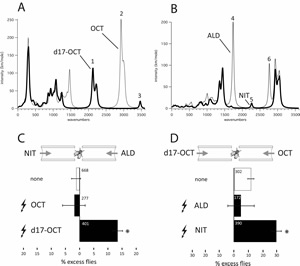 Olfaction presents the ultimate challenge to molecular recognition. Thousands of molecules are recognized by a small number of olfactory receptors and responses elicited from appropriate brain centers. Conventional molecular shape recognition mechanisms proposed to account for the exquisite odorant selectivity and discrimination exhibited by a limited number of olfactory receptors (~400 in humans, ~100 in the fly Drosophila melanogaster) can be stretched to combinatorial models to account for recognition of thousands molecules. However, the notorious lack of predictive power of the odor of particular molecules, exhibited by shape-based explanations led us to recent experiments which provide evidence for an alternative potentially complementary mechanism of olfactory recognition based on the molecular vibrations of odorant molecules. A plausible mechanism to explain the observations had been proposed in the 90’s by Turin. Empirically, this hypothetical mechanism can be used to predict the odor of particular molecules far better than shape-based theories. Therefore, based on recently published work, we propose that the Drosophila olfactory receptors likely utilize a biological form of inelastic electron tunneling - a quantum mechanism-to detect such molecular vibrations and thus may represent biological quantum sensors. Based on preliminary results we have proposed a molecular and a functional model for this sensor. We aim to dissect the sensor mechanism by functional genetic means, determine its selectivity by electrophysiology and study the theory and physics of tunneling electron transport in these olfactory receptors. Aside from the groundbreaking impact, results of this interdisciplinary proposal will have on the novel emerging field of Quantum Biology, a distinct tangible outcome of the proposed studies is reverse-engineering the fly quantum sensor.
Olfaction presents the ultimate challenge to molecular recognition. Thousands of molecules are recognized by a small number of olfactory receptors and responses elicited from appropriate brain centers. Conventional molecular shape recognition mechanisms proposed to account for the exquisite odorant selectivity and discrimination exhibited by a limited number of olfactory receptors (~400 in humans, ~100 in the fly Drosophila melanogaster) can be stretched to combinatorial models to account for recognition of thousands molecules. However, the notorious lack of predictive power of the odor of particular molecules, exhibited by shape-based explanations led us to recent experiments which provide evidence for an alternative potentially complementary mechanism of olfactory recognition based on the molecular vibrations of odorant molecules. A plausible mechanism to explain the observations had been proposed in the 90’s by Turin. Empirically, this hypothetical mechanism can be used to predict the odor of particular molecules far better than shape-based theories. Therefore, based on recently published work, we propose that the Drosophila olfactory receptors likely utilize a biological form of inelastic electron tunneling - a quantum mechanism-to detect such molecular vibrations and thus may represent biological quantum sensors. Based on preliminary results we have proposed a molecular and a functional model for this sensor. We aim to dissect the sensor mechanism by functional genetic means, determine its selectivity by electrophysiology and study the theory and physics of tunneling electron transport in these olfactory receptors. Aside from the groundbreaking impact, results of this interdisciplinary proposal will have on the novel emerging field of Quantum Biology, a distinct tangible outcome of the proposed studies is reverse-engineering the fly quantum sensor.