A role for reproductive tract development genes in Mayer-Rokitansky-Küster-Hauser syndrome and female infertility
The Dimas lab has led the Greek part of a multi-center study published in AJHG that addressed the genetic basis of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, a developmental syndrome leading to infertility in women. [Pubmed]
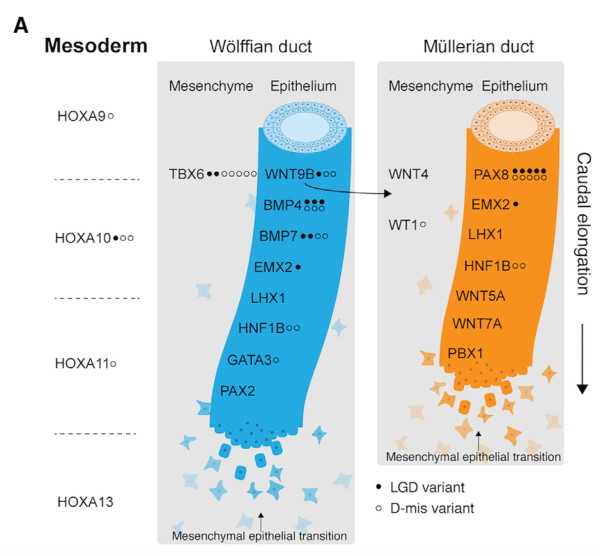
Genetic variants in seven genes were linked to the syndrome, with PAX8, a transcription factor expressed during embryonic development, representing the most significant MRKH-associated gene.
MRKH syndrome is a congenital disorder in which the Müllerian ducts fail to develop, leading to aplasia of the uterus and vagina. The syndrome affects 1/4,500 female live births, results in infertility, and may be accompanied by severe malformations of the urinary tract and spine. The genetic basis of MRKH was addressed through exome sequencing in a total of 592 cases from China, USA, Greece, Germany, France, Switzerland and Brazil.
The study was informed through the knowledge base of genitourinary developmental biology and 19 genes essential for Mullerian duct/Wolffian duct development were interrogated further. Following scanning for likely gene-disrupting (LGD) and deleterious missense (D-mis) variants, we report seven genes (PAX8, BMP4, BMP7, TBX6, HOXA10, EMX2, and WNT9B) harbouring genetic variants linked to the syndrome. Notably, sex-limited penetrance with paternal inheritance was observed in multiple families suggesting an inheritance mode for MRKH.
The findings add to our understanding of the development of the female reproductive system and of vital organs (e.g. kidneys) and also increase the value of genetic counselling offered to MRKH patients and family members. Importantly, this study also informs the way we think about developmental diseases and approaches that can be implemented to address their genetic basis.
Pubmed link: https://pubmed.ncbi.nlm.nih.gov/33434492/
PMID: 33434492