20/04/2017
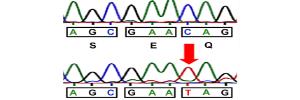
SLC25A46, a novel mitochondrial protein, has recently been identified as a pathogenic cause in a wide spectrum of rare human neurological diseases, including optic atrophy, Charcot-Marie-Tooth type 2, Leigh syndrome, progressive myoclonic ataxia and lethal congenital pontocerebellar hypoplasia. Following a genetic approach, Dr Douni's group identified a novel neurological mouse model caused by a functional mutation in the Slc25a46 gene. SLC25A46 is an outer membrane protein, member of the Solute Carrier 25 (SLC25) family of nuclear genes encoding mitochondrial carriers, with a role in mitochondrial dynamics and cristae maintenance. Mutant mice manifest the main clinical features identified in patients, including ataxia, optic atrophy and cerebellar hypoplasia, which were completely rescued by expression of the human ortholog. In collaboration with the research group of Dr Matsas, histopathological analysis revealed previously unseen lesions, most notably disrupted cytoarchitecture in the cerebellum and retina and prominent abnormalities in the neuromuscular junction. A distinct lymphoid phenotype was also evident. Our mutant mice provide a valid model for understanding the mechanistic basis of the complex SLC25A46-mediated pathologies, and further investigation of the associated human pathology.